Autor: Peter Pančík
Publikované dňa:
Citácia: PANČÍK, Peter. 2016. Biopedia.sk: Genetika človeka. [cit. 2024-04-20]. Dostupné na internete: <https://biopedia.sk/genetika/genetika-cloveka>.
Dedičnosť človeka nemožno skúmať z etického hľadiska metódou pokusného kríženia. Ľudské partnerské páry majú navyše málo potomkov na to, aby sa dedičnosť ktoréhokoľvek znaku mohla analyzovať priamo podľa štiepnych pomerov v jednotlivých generáciách. Pri priemernej generačnej dobe človeka (asi 27 rokov) môže genetik objektívne sledovať najviac 4 generácie. Každý človek je veľmi zložitým polyhybridom, má veľmi rozsiahly a individuálne zložito diferencovaný genotyp, a tým aj fenotyp. Človek žije v populáciách, ktoré sa odlišujú veľkosťou a mierou izolácie, v ktorých sa veľmi rozmanite uplatňujú vplyvy génového posunu.
Genetika človeka má teda v podstate k dispozícií namiesto experimentálnych metód pozorovacie metódy. Sleduje fenotypové prejavy osôb vybraných podľa určitého systému:
- genealogický výskum - skúma sa určitý rok niekoľko generácií
- populačný výskum - skúma sa náhodne vybratá vzorka populácie (výberový súbor)
- gemelologický výskum - skúmajú sa obidvaja jedinci páru dvojčiat
V praxi sa potom zvyčajne všetky tieto tri prístupy kombinujú, výsledky sa navzájom porovnávajú a dopĺňajú a získané údaje sa spracúvajú pomerne zložitými štatistickými metódami. V mnohých prípadoch treba vyšetrovať aj mikroskopický obraz chromozómov - karyotyp daného jedinca.
Cytogenetika link
Cytogenetika sa zaoberá štúdiom chromozómov (ich počtom, štruktúrou a segregáciou pri bunkovom delení) za normálnych a patologických podmienok a súvislosťou týchto nálezov s fenotypom. Chromozómy majú podľa stupňa špiralizácie odlišný morfologický vzhľad, inak vyzerajú v interfáze a inak v metafáze, kedy sú najhrubšie a najlepšie viditeľné. Pozornosť cytogenetiky sa upína na tieto fázy bunkového cyklu:
- profáza meiózy - sledovanie abnormalít počas párovania chromozómov
- metafáza - hlavne bunky ovplyvnené kolchicínom - tvorba karyotypu
- anafáza - sledovanie abnormalít počas rozchodu chromozómov
Počet, tvar a veľkosť chromozómov sú stále a druhovo špecifické znaky. Ako je známe, človek má v každej telovej bunke 46 chromozómov, tzn. 23 párov chromozómov. Jeden z týchto párov predstavuje gonozómy (chromozómy X a Y), zvyšné predstavujú autozómy. Kombinácia gonozómov určuje u ľudí pohlavie tak, že XX je žena a XY je muž. Všetky chromozómy sa podľa veľkosti a polohy centroméry rozdeľujú do 7 skupín označených A až G:
| skupina | autozómy | gonozómy | charakteristika chromozómov | počet u muža | počet u ženy |
| A | 1 - 3 | - | veľké viac-menej metacentrické | 6 | 6 |
| B | 4 - 5 | - | veľké submetacentrické | 4 | 4 |
| C | 6 - 12 | X | stredne veľké metacentrické | 15 | 16 |
| D | 13 - 15 | - | stredne veľké akrocentrické | 6 | 6 |
| E | 16 - 18 | - | malé submetacentrické | 6 | 6 |
| F | 19 - 20 | - | malé metacentrické | 4 | 4 |
| G | 21 - 22 | Y | malé akrocentrické | 5 | 4 |
Počet chromozómov nie je v žiadnom vzťahu s komplexnosťou organizmu alebo fylogenetickým postavením druhu.
| druh | počet chromozómov (2n) |
| drozofila (Drosophila melanogaster) | 8 |
| háďatko (Caenorhabditis elegans) | 12 |
| mucha domáca (Musca domestica) | 12 |
| arábovka (Arabidopsis thaliana) | 14 |
| hrach (Pisum sativum) | 14 |
| jačmeň (Hordeum vulgare) | 14 |
| raž (Secale cereale) | 14 |
| chlamydomonáda (Chlamydomonas reinhardtii) (n) | 17 |
| konope (Cannabis sativa) | 20 |
| kukurica (Zea mays) | 20 |
| ryža (Oryza sativa) | 24 |
| kvasinka (Saccharomyces cerevisiae) | 32 |
| mačka (Felis catus) | 38 |
| myš (Mus musculus) | 40 |
| sója (Glycine max) | 40 |
| pšenica (Triticum aestivum) | 42 |
| človek rozumný dnešný (Homo sapiens sapiens) | 46 |
| šimpanz (Pan troglodytes) | 48 |
| zemiak (Solanum tuberosum) (4n) | 48 |
| kôň (Equus caballus ferrus) | 64 |
| kur domáci (Gallus gallus) | 78 |
| pes (Canis lupus familiaris) | 78 |
| tur domáci (Bos taurus) | 78 |
| kapor (Cyprinus carpio) | 104 |
Monogénna dedičnosť u človeka link
Monogénne dedičnými nazývame také znaky, u ktorých jednotlivé fenotypové kategórie sú podmienené genotypom na jedinom lokuse a dedia sa klasickým mendelistickým spôsobom. Takto sa dedia aj krvné skupiny u človeka a mnoho dedičných chorôb, u ktorých sa dá štúdiom rodokmeňa určiť prognóza, tzn. pravdepodobnosť výskytu určitej poruchy u súrodencov a potomkov.
AB0 systém link
Krvná skupina je imunologický znak. Na krvnú skupinu nemá životné prostredie žiadny vplyv. Každá krvná skupina systému AB0 je monogénne dedičný znak a lokus príslušného génu je autozómový. Alely určujúce skupiny A a B sú úplne dominantné a navzájom kodominantné, tzn. že v heterozygotnom genotype sú vo fenotype vyjadrené obidve. Tretia alela pre krvné skupiny 0 je proti obidvom predchádzajúcim úplne recesívna. Znamená to, že lokus pre krvný systém AB0 je multialelický s tromi alelami IA, IB a i.
Krvná skupina A:
genotyp IAIA alebo IAi
Krvná skupina B:
genotyp IBIB alebo IBi
Krvná skupina AB:
genotyp IAIB
Krvná skupina 0:
genotyp ii
Molekulárna podstata krvných skupín link
Veľké množstvo antigénov, medzi ktoré patria aj aglutinogény nachádzajúce sa na vonkajšej strane plazmatickej membrány červených krviniek, sú charakteru oligosacharidov, resp. glykolipidov. Ich špecifickosť je určená prítomnosťou konkrétnych monosacharidových zvyškov a ich usporiadaním (vetvenie oligosacharidového reťazca). Za tvorbu krvných skupín sú zodpovedné dve glykozyltransferázy, čo sú enzýmy, ktoré sa podieľajú na prenose špecifického monosacharidu na základnú oligosacharidovú "kostru" (alela i nepridáva žiadny monosacharid):
Krvná skupina A:
alela: IA
prenos monosacharidu: N-acetylgalaktozamín
Krvná skupina B:
alela: IB
prenos monosacharidu: galaktóza
Krvná skupina 0:
alela: i
prenos monosacharidu: -

Rh faktor link
Rh-faktor je príkladom klasického autozómového typu dedičnosti, tzn. že dominantná alela Rh+ určuje Rh-pozitívnu krv, recesívna alela Rh- určuje Rh-negatívnu krv.
Príklad na dedičnosť krvných skupín link
Príklad: Klasický príklad na dedičnosť krvných skupín: Aká je pravdepodobnosť, že matka s krvnou skupinou A+ a otec so skupinou B+budú mať dieťa 0-?
Poznáme krvné skupiny obidvoch rodičov, presne však nepoznáme ich genotypy ( IA-Rh+-, IB-Rh+-). Ak však poznáme skupinu dieťaťa s jasným genotypom (iiRh-Rh-), môžeme dedukovať, že v tomto prípade môžu mať dieťa so skupinou 0- len rodičia heterozygotní v obidvoch znakoch ( IAiRh+Rh-, IBiRh+Rh-). Zápis kríženia vyzerá teda nasledovne:
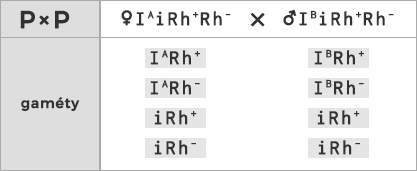
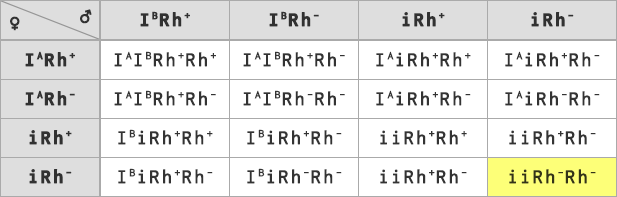
Odpoveď: Pravdepodobnosť, že dieťa bude mať krvnú skupinu 0- je 1 : 16 (6,25%).
Genetické choroby link
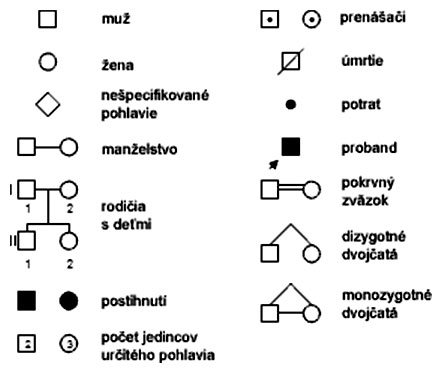
Geneticky podmienené patologické stavy predstavujú u človeka závažný medicínsky problém. Ich prejav prakticky nesúvisí s vplyvom vonkajšieho prostredia, pretože vychádza zo samotnej poruchy genetickej informácie, ktorá je prítomná v každej bunke ľudského tela. Prognózu ochorenia pre súrodencov a ďalšie generácie možno určiť pomocou zostavenia rodokmeňa, ktorý obsahuje údaje týkajúce sa študovaného znaku v príbuzenstve (genealógia). Zostavuje sa na požiadanie a na základe probanda, čo je osoba, u ktorej bola genetická choroba prvýkrát zaznamenaná. V súvislosti s rodokmeňmi rozlišujeme niekoľko genealogických symbolov:
Príčinou genetických porúch, ktoré sa vyskytujú u živonarodených detí, sú génové mutácie špecifických génov. Ich podstatou je malá zmena genetickej informácie, preto sa nazývajú aj bodové mutácie. Mutácia spôsobí neschopnosť vytvárať alebo v nedostatočnom množstve vytvárať príslušný polypeptidový reťazec (bielkovinu), ktorý má často funkciu enzýmu alebo hormónu. Organizmus teda nemá schopnosť určitej špecifickej metabolickej alebo fyziologickej reakcie. Podstatou takých chorôb je, že chýba určitý typ bielkovinovej molekuly, nazývame ich preto molekulovými chorobami.
Dedičné choroby z hľadiska naviazanosti na chromozómy môžu byť:
- autozómové - viazané na nepohlavný chromozóm,
- gonozómové - viazané na pohlavný chromozóm X alebo Y,
...pričom na základe vzťahu medzi alelami na danom lokuse sú dvojakého charakteru:
- dominantné - ochorejú aj heterozygoti,
- recesívne - ochorejú iba homozygoti pre príslušnú mutovanú alelu.
Autozómovo dominantná dedičnosť (AD) link
Ak je dominantná alela D v populácii veľmi zriedkavá, ako je to často prípad genetických chorôb, je takmer isté, že jedinec dominantného fenotypu je heterozygot Dd, a teda všetky manželstvá sú typu Dd × dd. Jedná sa predovšetkým o mutácie základných bielkovín morfologicko-štruktúrneho charakteru, bielkovín vo funkcii biologických nosičov a receptorov bunky, zriedka nimi bývajú bielkoviny enzýmového charakteru.
Typickými znakmi autozómovo dominantne dedičného znaku v rodokmeni sú:
- vertikálny prenos v rodokmeni (postihnuté dieťa má postihnutého rodiča) cez viac generácií
- pravdepodobnosť postihnutia potomka postihnutého rodiča 0,5
- pravdepodobnosť postihnutia potomka ak sú obaja rodičia postihnutí 0,75
- podiel postihnutých žien a mužov je rovnaký
- prenos do ďalšej generácie cez obidve pohlavia
Niekedy je komplikáciou pri hodnotení typu dedičnosti AD poruchy variabilná expresivita a neúplná penetrancia znaku. V prvom prípade sa jedná o rozličný kvalitatívny a kvantitatívny prejav znaku u jedincov s rovnakým genotypom. Druhý prípad súvisí s prejavom znaku u jedincov s rovnakým genotypom len v niekoľkých percentách prípadov, príp. so začiatkom prejavov v odlišnom veku. Ak sa choroba prejaví vždy, jedná sa o 100% penetranciu. Najčastejšou príčinou oboch stavov je heterozygotná konštitúcia (typická pre AD choroby), vplyv iných génov alebo prostredia. Oba termíny tak v podstate vyjadrujú len naše nedostatočné vedomosti o etiológii a spôsobe dedičnosti daného znaku.
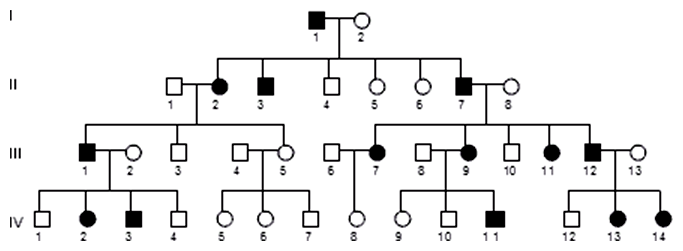
Veľmi zriedkavým (1 : 1 000 000) dominantne autozómovým dedičným ochorením je familiárna hypercholesterolémia. Jedinci s týmto dedičným ochorením majú mutáciu v géne pre LDL receptor (špecifický bielkovinový receptor na lipoproteínovom komplexe, ktorý prenáša cholesterol v krvi). Dôsledkom tejto poruchy je neschopnosť periférnych tkanív prijímať cholesterol z krvi, cholesterol sa hromadí v krvnom obehu a stáva sa vážnym rizikovým faktorom vzniku artériosklerózy a srdcového infarktu.
Autozómovo recesívna dedičnosť (AR) link
Aj pri tomto prípade dedičnosti genetickej choroby pochádza väčšina postihnutých z krížení dvoch heterozygotov Dd × Dd, pretože alela je v populácii pomerne zriedkavá. Produktom patologického génu je predovšetkým enzýmový defekt. Variabilita stupňa závažnosti patologického stavu je zvyčajne slabšie vyznačená ako pri dominantnej autozómovej dedičnosti.
Typickými znakmi autozómovo recesívne dedičného znaku v rodokmeni sú:
- postihnutí zvyčajne len v jednej generácii (horizontálny prenos)
- rodičia postihnutých sú zvyčajne zdraví
- pravdepodobnosť postihnutia ďalšieho súrodenca postihnutého je 0,25 bez ohľadu na počet postihnutých v súrodenectve
- postihnutí majú zvyčajne zdravých potomkov
- podiel postihnutých žien a mužov je rovnaký
- častejší výskyt príbuzenských sobášov u rodičov
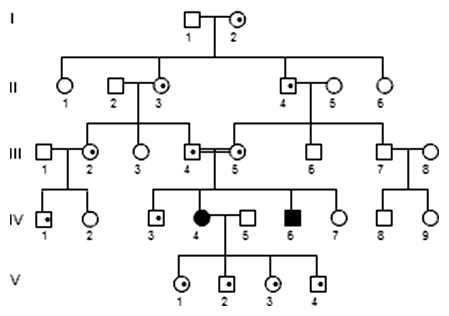
Príkladom recesívne autozómového dedičného ochorenia je galaktozémia, pri ktorej je postihnutý organizmus neschopný vytvárať jeden z enzýmov nevyhnutných na normálny priebeh katabolického reťazca odbúravania mliečneho cukru - galaktózy. Metabolická dráha teda prebieha až po určitý medziprodukt, tu sa zastaví a ďalej nespracovateľný medziprodukt sa hromadí vo väčšine vnútorných orgánov, na ktoré pôsobí ako jed. Vznikajú tak vážne príznaky: nechutenstvo a iné tráviace ťažkosti, znížená hladina glukózy v krvi, žltačka, zmrštenie pečene, zväčšenie sleziny, príznaky poškodenia obličiek, zákal očnej šošovky a spomalenie psychického vývinu. Genetická informácie jednej mutovanej alely sa teda premieta do mnohých patologických znakov - pleiotropný účinok. Vzniku tejto choroby možno predísť vylúčením mlieka a mliečnych výrobkov zo stravy.
Gonozómovo dominantná dedičnosť (XD) link
Dominantnými genetickými chorobami, ktoré sú viazané na X-chromozóm, sú postihnuté aj ženy, avšak často je prejav ochorenia miernejší. Je to pravdepodobne preto, že sú zvyčajne heterozygotky XDXd a ako u všetkých buniek s karyotypom XX, dochádza v prvých štádiách ontogenézy k inaktivácii jedného X-chromozómu (Lyonovej hypotéza inaktivácie jedného X-chromozómu u žien). Pritom je často inaktivovaný práve X-chromozóm s defektnou alelou.
Typickými znakmi gonozómovo dominantne dedičného znaku v rodokmeni sú:
- sú postihnutí muži aj ženy
- podiel postihnutých žien v populácii je zhruba 2x vyšší
- potomok postihnutej ženy má 50% riziko postihnutia bez ohľadu na pohlavie
- postihnutý muž má všetky dcéry postihnuté a žiadneho postihnutého syna
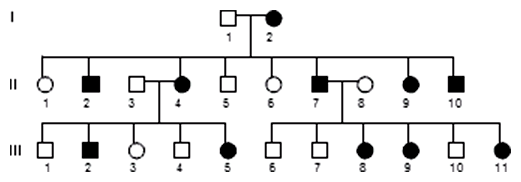
V praxi je známych málo prípadov genetických chorôb s týmto typom dedičnosti.
Gonozómovo recesívna dedičnosť (XR) link
Matka je zvyčajne asymptomatická prenášačka a môže mať postihnutých potomkov mužského pohlavia. Postihnutie žien môže nastať podobne ako v predchádzajúcom prípade inaktiváciou chromozómu, ktorý nesie "zdravú" alelu. Taktiež ženy postihnuté Turnerovým syndrómom (karyotyp 45,X0) majúce len jeden X-chromozóm sú v podobnej pozícii ako hemizygotný muž (46,XY), takže sa u nich recesívna alela môže prejaviť.
Typickými znakmi gonozómovo recesívne dedičného znaku v rodokmeni sú:
- zvyčajne postihnutí sú len muži
- postihnutí muži sa rodia nepostihnutým rodičom
- postihnutí muži neprenášajú ochorenie na svojich synov
- všetky dcéry postihnutých mužov sú prenášačky
- môže sa vyskytovať preskočenie generácie
- nie je zvýšený výskyt konsangvinity (pokrvných sobášov) medzi rodičmi postihnutých

Najčastejšia recesívne gonozómová porucha krvnej zrážanlivosti novonarodených chlapcov (1 : 5000-10 000) je klasická hemofília A, ktorej gén sa nachádza na X-chromozóme. U chlapcov, keďže majú len jeden X-chromozóm, dôjde vždy k postihnutiu, u dievčat, ktoré majú X-chromozómy dva, choroba sa neprejaví (len v prípade homozygotne recesívneho stavu). Dievčatá s jednou zmutovanou alelou sú prenášačky a môžu hemofíliu preniesť na svoje potomstvo.
Iným častým príkladom recesívne gonozómovej dedičnej poruchy viazanej na X-chromozóm je daltonizmus - farbosleposť na zelenú a červenú farbu, ktoré postihnutý jedinec vníma ako rôzne odtiene šedej farby.
Holandrická dedičnosť (Y) link
Je špeciálnym typom dedičnosti znakov, ktoré sa nachádzajú na Y-chromozóme. Zatiaľ nebol zistený žiadny patologický znak, ktorý by sa prenášal touto cestou.
Typickými znakmi holandrickej dedičnosti znaku v rodokmeni sú:
- postihnutí sú len muži
- všetci synovia postihnutých mužov sú tiež postihnutí
Chromozómové aberácie link
Človek má v somatických bunkách 23 párov chromozómov (46 chromozómov), z čoho je 22 párov autozómov a 1 pár gonozómov (sexozómov), ktoré sú heterologické a označujú sa X a Y. Pohlavné bunky majú po jednom chromozóme z každého chromozómového páru, teda celkom 23 chromozómov. Heterogametické pohlavie (XY) je u človeka (a drozofily) mužské (samčie), homogametické pohlavie (XX) je ženské (samičie). Na chromozómoch je DNA usporiadaná lineárne a jej jednotlivé úseky sú uložené v charakteristických väzbových skupinách, tvoriac tak komplexnú genetickú informáciu organizmu.
Zmeny v počte alebo štruktúre chromozómov majú tak vážne dôsledky na prežívanie jedinca. Napriek tomu existujú patologické prípady zlučiteľné so životom. Ich reprodukčná schopnosť je však do značnej miery potlačená, pretože buď sa daný jedinec reprodukčného veku nedožije alebo je znemožnená tvorba životaschopných pohlavných buniek počas gametogenézy. Chromozómovými aberáciami sú charakteristické prevažne ľudské nádory.
Niektoré genetické choroby súvisiace so zmenou štruktúry chromozómov:
- syndróm Cri du chat (syndróm "mačacieho plaču") - delécia časti krátkeho ramena chr. 5
- Praderov-Williho syndróm - delécia časti dlhého ramena chr. 15
- chronická myeloidná leukémia - reciproká translokácia medzi chr. 9 a 22
- Burkittov lymfóm - reciproká translokácia medzi chr. 8 a 14
- fragilný chromozóm X - mnohonásobná duplikácia DNA na dlhom ramene chr. X
- myotonická dystrofia - mnohonásobná duplikácia DNA na dlhom ramene chr. 19
V súvislosti so zmenou počtu chromozómov poznáme predovšetkým trizómie, ktoré sú zlučiteľné so životom. Monozómie ako aj polyploidie vedú k potratu v skorých štádiách ontogenézy. Najmenej závažné sú aneuploidie spojené s pohlavnými chromozómami, u ktorých je u žien tolerovaná aj monozómia a tetrazómia:
- Downov syndróm - trizómia chr. 21
- Patauov syndróm - trizómia chr. 13
- Edwardsov syndróm - trizómia chr. 18
- Turnerov syndróm - monozómia chr. X
- "superžena" - trizómia, tetrazómia alebo pentazómia chr. X
- "supermuž" - karyotyp XYY
- Klinefelterov syndróm - karyotypy XXY, XXYY, XXXY
Inbríding link
Inbríding predstavuje uzatváranie manželstiev medzi geneticky príbuznými jednotlivcami (majú aspoň jedného spoločného predka, ktorý nie je vzdialenejší ako 4-5 generácií). Rozlišujeme dva druhy inbrídingu:
- systematický inbríding - príbuzenské maželstvá s definovaným typom príbuznosti (napr. bratranec-sesternica)
- nesystematický inbríding - manželstvá v rámci malej geneticky izolovanej populácie
Genetické poradenstvo link
Spoločnou úlohou genetiky a medicíny je včas a správne spoznať a liečiť choroby s dedičnou dispozíciou a dedičné choroby. Okrem toho je ich úlohou správne predpovedať možnosť ďalšieho výskytu týchto chorôb v rodinách a rodoch, kde sa už vyskytli, a na základe genetickej prognózy včas odporučiť účinné preventívne opatrenia. Usilujú sa aj o to, aby sa vážne dedičné choroby neprenášali na ďalšie generácie, teda snažia sa predísť splodeniu potomkov, ktorí by nežiadúce znaky dedili a prenášali ďalej.
V každom kraji pracuje Genetické oddelenie krajského ústavu národného zdravia. Ich hlavnou úlohou je genetické poradenstvo, registrácia všetkých dedičných chorôb a lekársko-genetický výskum. Najvýznamnejšia a pomerne samostatná zložka každého oddelenia je Genetická poradňa. Lekári-genetici sa musia presne oboznámiť s klinickou a genetickou situáciou príslušného prípadu a na vedeckom základe potom stanoviť genetickú predpoveď (prognózu) - pravdepodobnosť ďalšieho výskytu danej dedičnej choroby alebo chyby.
Cieľom tejto činnosti je genetická prevencia, t.j. predchádzanie dedičnému zaťaženiu ďalších ľudí: činnosť genetických poradní je konkrétnym prejavom eugenických snáh v ľudskej spoločnosti. Eugenika je hraničný odbor geneticko-lekársko-sociologický, ktorý využíva poznatky a možnosti všetkých troch vied v prospech zlepšovania génového fondu (genofondu), a tým aj dedičných znakov ľudstva.