
I. generácia sekvenovania link
Odkedy FREDERICK SANGER (1918–2013) objavil v roku 1977 enzymatickú metódu sekvenovania DNA založenú na použití dideoxy-nukleotidov, stala sa štandardom molekulárno-biologického výskumu na nasledujúcich 30 rokov. Počas tohto obdobia dochádzalo k mnohým technickým vylepšeniam, k zvyšovaniu bezpečnosti či citlivosti. Tomuto rozvoju do veľkej miery napomohol Projekt sekvenovania ľudského genómu (angl. Human Genome Project, HGP), ktorý prebiehal v rokoch 1990 až 2003. Trval 13 rokov, stál približne 3 miliardy dolárov a jeho výsledkom bolo osekvenovanie 99 % ľudského genómu o veľkosti 3,3 miliardy bázových párov.
Významným krokom rozvoja bolo zavedenie automatizácie s minimálnymi požiadavkami na personál. V roku 1996 uviedla firma Applied Biosystems na trh prvý komerčný automatický sekvenátor. Avšak ani ďalší vývoj týchto prístrojov nedokázal dosiahnuť vtedajší stanovený cieľ: sekvenovať celý ľudský genóm pod hranicu 1000 dolárov. Kapacita jednoducho narážala na svoje fyzikálne limity, keďže v jednom behu bolo možné sekvenovať maximálne 384 rôznych sekvencií s dĺžkou do 1000 bp.
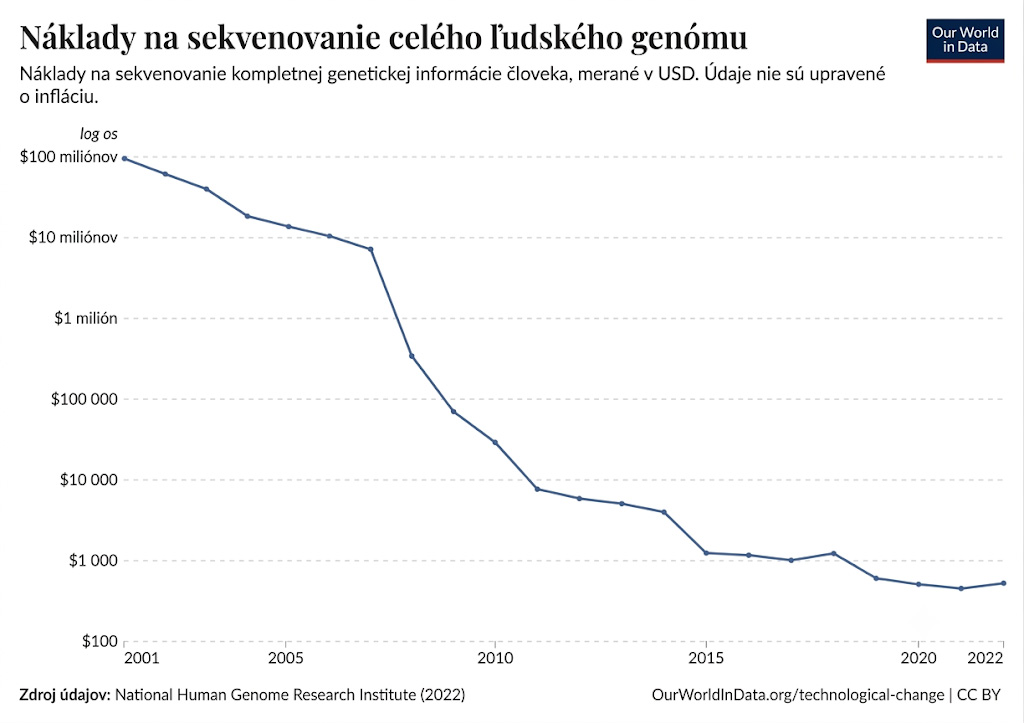
Odhaduje sa, že čítanie celého ľudského genómu klasickou metódou by aj v súčasnosti stálo desiatky miliónov dolárov a trvalo by niekoľko rokov.
Obdobie monopolu klasického Sangerovho sekvenovania sa nazýva aj I. generácia sekvenovania a trvalo približne do roku 2005. Po roku 2005 však prebrali štafetu technológie sekvenovania novej generácie (NGS), vďaka ktorým klesla cena sekvenovania ľudského genómu z 5 800 dolárov v roku 2012 na úroveň približne 500 dolárov (podľa dát z roku 2022), pričom samotný proces dnes trvá len niekoľko dní. Úmerne k tomu enormne vzrástlo aj množstvo novo-osekvenovaných genómov na státisíce ročne.
II. generácia sekvenovania link
Zatiaľ čo obdobie po roku 2005 sa historicky označovalo ako II. generácia sekvenovania, s príchodom a rozvojom ďalších prístupov sa dnes vžil skôr zastrešujúci pojem sekvenovanie novej generácie (NGS, angl. Next-Generation Sequencing). Z technologického hľadiska sa dnes NGS rozdeľuje na dva základné smery:
- sekvenovanie s krátkymi čítaniami (angl. short-read sequencing) – historicky označované ako II. generácia, kde sa masívne paralelne čítajú obrovské množstvá krátkych úsekov DNA (desiatky až stovky bázových párov)
- sekvenovanie s dlhými čítaniami (angl. long-read sequencing) – označované aj ako III. generácia, schopné čítať súvislé úseky dlhé tisíce až desiatky tisíc bázových párov bez nutnosti ich predchádzajúcej amplifikácie (tomuto prístupu sa venuje samostatná kapitola)
Za prielom v sekvenovaní s krátkymi čítaniami sa považuje obdobie 2005–2007, kedy hneď niekoľko firiem prinieslo na trh nové technológie. Tie umožnili sekvenovať veľké úseky DNA za oveľa menej peňazí a v podstatne kratšom čase:
- GS (Genome Sequencer) FLX System od firmy Roche (r. 2005, pôvodne 454 pyrosequencer od 454 Life Sciences)
- Illumina Genome Analyzer od firmy Applied Biosystems (r. 2006, pôvodne Solexa 1G)
- SOLiD DNA Sequencer (r. 2007) od firmy Applied Biosystems
- Ion Torrent (r. 2010) od firmy Life Technologies
Spoločnou charakteristikou tejto tzv. II. generácie (short-read NGS) je použitie mikro- a nanotechnológií, ktoré umožnili minimalizáciu množstva vzorky a všetkých komponentov reakcie, a zároveň masívne paralelné sekvenovanie veľkého množstva DNA sekvencií v jednom momente. Na rozdiel od Sangerovho sekvenovania produkujú podstatne kratšie reťazce (klasický Sanger do 1000 bp vs. moderná Illumina zvyčajne do 150 až 250 bp, Ion Torrent 200–400 bp).
Zatiaľ čo klasické Sangerovo sekvenovanie prebieha na templáte, ktorým je jednoduchý PCR produkt alebo purifikovaná plazmidová DNA, príprava templátu pre short-read NGS je samostatným krokom, ktorý je rovnako dôležitý ako samotné sekvenovanie. Keďže princípom tejto metódy je paralelné sekvenovanie obrovského množstva vzoriek, templát je tvorený mnohými rôznymi fragmentmi DNA upravenými tak, aby sa dali čítať prístrojom. Takýto súbor sekvencií sa zjednodušene nazýva DNA knižnica.(1)(2)(3)(4)

Celkovo tak postup short-read sekvenovania pozostáva zo 4 základných krokov:
- príprava DNA templátu, resp. DNA knižnice
- amplifikácia (namnoženie) DNA knižnice (tento krok je špecifický práve pre krátke čítania, aby sa zosilnil signál pre prístroj)
- samotné sekvenovanie
- bioinformatická analýza dát
Tvorba DNA knižnice link
Genomická alebo akákoľvek DNA sa pred sekvenovaním musí posekať (fragmentovať) na menšie kúsky (do 500–1000 bp), ktoré sa sekvenujú samostatne. Tieto fragmenty sa potom prichytávajú buď na pevnú podložku (Illumina) alebo na magnetické nanoguličky (historicky technológie 454 a SOLiD, dnes Ion Torrent). Knižnica môže mať lineárny (Illumina) alebo kruhový tvar (Ion Torrent, a v minulosti 454, SOLiD).
Amplifikácia DNA knižnice link
Amplifikácia DNA knižnice sa deje ešte pred samotným sekvenovaním z dôvodu zvýšenia detekčného signálu, ktorý vie poskytnúť len pomnožená DNA v mnohých kópiách. Amplifikácia prebieha priamo na mieste, v emulzii (emulzná PCR, emPCR – dnes Ion Torrent, v minulosti 454, SOLiD) alebo priamo na pevnom povrchu (Illumina). Ak je na hybridizáciu DNA fragmentov použité pevné sklíčko (tzv. flow cell), pripravená knižnica je rozdelená na veľké množstvo zhlukov (angl. clusters) (Illumina), ktoré sa sekvenujú ako samostatné jednotky. Podobne, jedna nanogulička (Ion Torrent, 454, SOLiD) predstavuje samostatnú sekvenačnú jednotku.
Sekvenovanie link
Sekvenovanie je založené na princípe syntézy komplementárneho vlákna z každého DNA fragmentu (Illumina, Ion Torrent, staršie 454) alebo ligácie 8-bázových prób (staršie SOLiD). Ide o masívne paralelné sekvenovanie celej knižnice. Každý zo štyroch voľných deoxy-nukleotidov (dNTP) je spravidla značený inou fluorescenčnou farbičkou (ako je to u dominujúcej Illuminy, staršie aj značené próby u SOLiD), alebo sekvenovanie prebieha cyklickým pridávaním a odmývaním jednotlivých nukleotidov (Ion Torrent, staršie 454). Po zaradení (inkorporovaní) nukleotidu alebo próby, dôjde k zaznamenaniu signálu z každého klastra, resp. nanoguličky. Pri Illumine (a staršom SOLiD) ide o detekciu fluorescencie, pri systéme 454 išlo o detekciu svetla (pyrosekvenovanie) a pri Ion Torrent o detekciu zmeny pH. Toto sa cyklicky opakuje, až kým nie je osekvenovaná celá knižnica.

fragment A: ...GAG...
fragment B: ...ATC...
fragment C: ...TAC...
Analýza sekvenačných dát link
Amplifikácia DNA knižnice a samotné sekvenovanie prebieha automaticky, bez zásahu užívateľa. Odhliadnuc od rôznej technickej a časovej náročnosti prípravy DNA knižnice, je samotné vyhodnotenie obrovského množstva dát vždy náročné a vyžaduje si nemalé vedomosti z oblasti bioinformatiky. Najmä v prípade, ak sa sekvenuje neznáma DNA (tzv. de novo sekvenovanie), ku ktorej zatiaľ neexistuje nijaká známa, príbuzná sekvencia (tzv. referenčná sekvencia), bolo skladanie výsledného genómu o veľkosti mnoho megabáz z krátkych fragmentov do 200 bp mimoriadne prácne. Spracovanie gigabajtov dát je v neposlednom rade náročné aj na technické vybavenie (výkon počítača) a čas.
Pyrosekvenovanie link
Pyrosekvenovanie je technológia II. generácie sekvenovania, pôvodne vyvinutá firmou 454 Life Sciences v roku 2005 a inkorporovaná do komerčne dostupných pyrosekvenátorov rady 454 (neskôr ju kúpila firma Roche a integrovala do prístrojov rady GS FLX System).
Na svoju dobu bolo obrovskou výhodou 454 technológie dlhé čítanie (až do 1000 bp s presnosťou 99,9 %) a na vtedajšie pomery veľká rýchlosť. Postupom času sa však stala jej osudnou mimoriadne pracná príprava DNA knižnice (emPCR) a príliš vysoká cena sekvenovania. Tieto faktory viedli k jej definitívnemu nahradeniu lacnejšími a výkonnejšími technológiami.

Príprava DNA knižnice a amplifikácia link
Vzorka DNA sa najprv fragmentuje na úseky dlhé 300–500 bp, ku ktorým sa z oboch strán pripoja špecifické adaptory (A a B). Tieto úseky sa následne prichytia na magnetické nanoguličky tak, aby na každú guličku pripadol štatisticky presne jeden fragment DNA. Amplifikácia (namnoženie fragmentov) prebieha v takzvanej emulznej PCR (emPCR) – v kvapôčkach oleja, ktoré zabezpečia, že každá gulička funguje ako samostatný a izolovaný „reaktor“.
Po namnožení sa guličky nanesú na špeciálnu pikotitračnú platničku, ktorej miniatúrne jamky sú rozmerovo prispôsobené tak, aby do každej z nich zapadla práve jedna nanogulička.(5)(6)(7)
Princíp sekvenovania (chemizmus) link
Chemizmus pyrosekvenácie je cyklický a pomerne zložitý. Nevyužíva fluorescenčne značené nukleotidy; rozpoznávanie poradia báz v DNA je dané tým, že do reakcie sa postupne a oddelene pridáva vždy len jeden zo štyroch voľných nukleotidov (dCTP, dGTP, dTTP alebo dATP). Aby sa predišlo falošným signálom, namiesto klasického dATP sa používa jeho modifikovaná verzia dATPαS.

Celý detekčný proces v jamke prebieha ako kaskáda štyroch enzýmov:
- DNA-polymeráza – ak je pridaný nukleotid komplementárny k predlohe, polymeráza ho zaradí do novovznikajúceho reťazca DNA. Pri tejto reakcii sa odštiepi molekula pyrofosfátu (PPi).
- ATP sulfuryláza – tento enzým okamžite spotrebuje uvoľnený pyrofosfát a za prítomnosti substrátu APS (adenozín-5-fosfosulfát) z neho vytvorí energeticky bohaté ATP.
- luciferáza – využíva novovzniknuté ATP ako „palivo“ na premenu látky luciferín na oxyluciferín. Táto reakcia produkuje záblesk svetla, ktorý prístroj zaznamená ako signál (peak) na výstupnom grafe (pyrograme). Intenzita signálu je priamo úmerná množstvu vytvoreného ATP – ak sa do sekvencie zaradia dva rovnaké nukleotidy za sebou, vznikne dvojnásobné množstvo ATP a záblesk je dvojnásobne silný.
- apyráza – na konci cyklu tento enzým degraduje zvyšné ATP a nespotrebované nukleotidy. Tým sa svetelný signál vypne, cyklus sa „reštartuje“ a prístroj môže pridať ďalší typ nukleotidu.

Illumina link
Illumina je technológia, ktorú pôvodne vyvinula firma Solexa a následne ju odkúpila a spopularizovala rovnomenná spoločnosť Illumina. Historicky bola táto technológia reprezentovaná prístrojmi ako Genome Analyzer, HiSeq, HiScanHQ a MiSeq.(8)

Príprava DNA knižnice pri systéme Illumina je principiálne podobná staršiemu systému 454, ale odlišný je nosič jednovláknových DNA fragmentov, ktorým je sklenená platnička s predhybridizovanými primermi (obr., B). Tieto primery sú komplementárne k adaptorovej sekvencii DNA fragmentov (obr., A). Hybridizácia DNA fragmentov bola pri prvých generáciách prístrojov priestorovo náhodná, pričom veľmi dôležitá bola ich primeraná hustota.
Po naviazaní DNA fragmentu na platničku (obr., B) dochádza k jeho ohybu a hybridizácii jeho voľného konca s adaptorom k predhybridizovanému primeru na platničke, takže pripomína most (angl. bridge) (obr., C). Následne prebehne amplifikácia (tzv. bridge-PCR) priamo na platničke s využitím tohto primeru, čím vznikne „dvojvláknový most“ (obr., D). Po následnej denaturácii sa štruktúra rozpadne (vlákna sa „vystrú“), ale už sú prítomné 2 jednovláknové molekuly, uchytené na platničke (obr., E). Takto postupne vznikne niekoľko 1000 kópií daného DNA fragmentu v tesnej blízkosti, čím sa vytvorí klaster (obr., F), funkčne ekvivalentný jednej nanoguličke systému 454 či Ion Torrent.
Samotné sekvenovanie sa však od iných platforiem odlišuje; ide o masívne paralelné sekvenovanie syntézou spojené s reverzibilnou termináciou polymerizácie (obr., I, J). Mastermix obsahuje primery, DNA-polymerázu a 4 rôzne fluorescenčne značené nukleotidy s reverzibilne odštiepiteľným terminátorom na 3'-konci, ktoré prechodne zastavujú polymerizáciu (fungujú podobne ako dideoxy-nukleotidy pri Sangerovom sekvenovaní). Po naviazaní týchto nukleotidov sa platnička premyje (odstránia sa nenaviazané nukleotidy) a laser zaznamená fluorescenciu podľa typu nukleotidu (obr., G, H, I), resp. fluorescenčnej farbičky (A, C, T, G). Napokon sa terminátor z 3'-konca odštiepi (obr., I, J) a do reakcie znovu vstupujú jednotlivé komponenty, ktoré zahája ďalšiu polymerizáciu a ďalšie meranie fluorescencie.
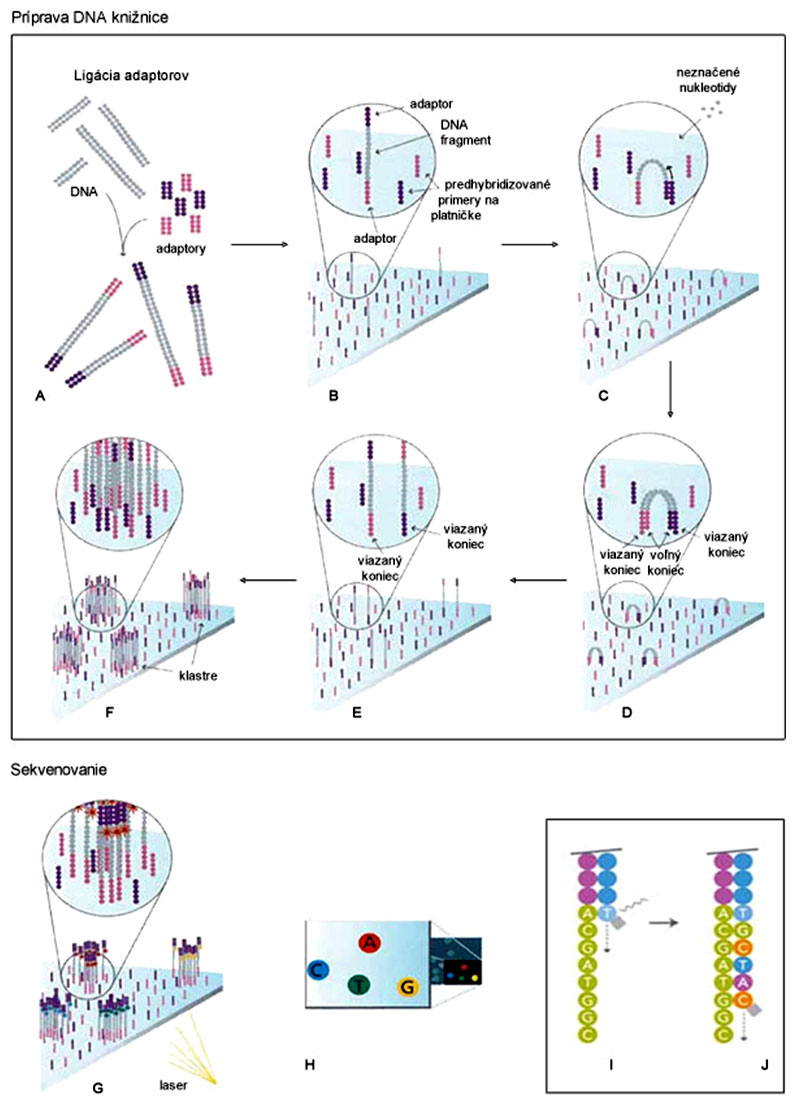
Najväčšou výhodou Illuminy je cena sekvenácie, ktorá sa historicky pohybovala na úrovni 0,07 $ / 1 megabázu. Nevýhodou bolo krátke čítanie (DNA fragmenty do 150 bp) a nižšia presnosť, predovšetkým na konci behu.
Ion Torrent link
Ion Torrent je technológia, ktorú v roku 2010 komercionalizovala firma Life Technologies (dnes celá táto technológia spadá pod korporáciu Thermo Fisher Scientific). Hlavnou funkčnou jednotkou prístrojov (historicky Ion PGM, Ion Proton) je polovodičový čip, ktorý rozhoduje o výkone a kapacite sekvenovania. Čip je zároveň priamym detektorom, ktorý prenáša chemickú informáciu, zakódovanú v A, C, T, G, na digitálnu informáciu (1, 0) v závislosti od prítomnosti, resp. neprítomnosti konkrétneho nukleotidu v danom cykle.(9)

DNA knižnica je fixovaná na nanoguličku a vzniká amplifikáciou DNA fragmentov pomocou emPCR. Signál zaznamenáva prístroj prostredníctvom jemnej zmeny pH, spôsobenej vylúčením protónu (H⁺) po úspešnej inkorporácii nukleotidu pri syntéze vlákna. Ide teda, podobne ako v predchádzajúcich technológiách, o sekvenovanie syntézou. Podobne, ako v prípade staršieho systému 454, zaradenie dvoch za sebou idúcich rovnakých nukleotidov v jednom cykle vedie k dvojnásobnému vylúčeniu H⁺ a zaznamenaný je výraznejší peak. Rozlíšenie nukleotidov (A, C, T, G) je dané cyklickým pridávaním a následným odmývaním jednotlivých voľných dNTP do reakcie.
Výhodou Ion Torrentu je jednoznačne rýchlosť a jednoduchosť: ide o priamu, chemickú detekciu bez potreby skenovania, drahých kamier alebo svetla. Historickou nevýhodou bola relatívne vyššia cena (v minulosti okolo 1 $ / 1 megabázu) a kratšie čítania pri porovnaní s nastupujúcimi konkurentmi.
SOLiD link
SOLiD (angl. Sequencing by Oligonucleotide Ligation and Detection) je technológia pôvodne vyvinutá firmou Applied Biosystems. Na rozdiel od predchádzajúcich technológií nevyužívala sekvenovanie syntézou, ale sekvenovanie ligáciou sond (prób) hybridizovaných k DNA knižnici.

Príprava knižnice a princíp ligácie link
DNA knižnica je amplifikovaná emulznou PCR na nanoguličkách, ktoré sú nanesené na sklenenú podložku.(10) Najprv hybridizuje univerzálny primer k adaptorovej sekvencii a následne k nemu enzým ligáza pripojí sekvenčne špecifickú sondu s naviazanou fluorescenčnou farbičkou na 5'-konci. Sonda je oktamér (8 nukleotidov), ktorý obsahuje 2 informatívne nukleotidy na 4.–5. pozícii (alebo 1.–2. pri novších verziách, ako je na obrázku nižšie). Zvyšných 6 nukleotidov tvoria univerzálne (z) alebo degenerované bázy (n), ktoré sa párujú s akýmkoľvek nukleotidom. Štyri fluorescenčné farbičky (FAM, CY3, TXR, CY5) určujú pomocou premysleného farebného kódu (ColorSpace) všetkých 16 možných dinukleotidov, pričom k jednej farbičke prislúchajú vždy 4 dinukleotidové kombinácie.

Priebeh sekvenovania a reštart cyklov link
Po ligácii sondy dochádza k čítaniu fluorescencie pomocou lasera (obr., A) a následnému odštiepeniu troch univerzálnych báz (vrátane fluorescenčnej farbičky) z 5'-konca sondy (obr., B). Cyklus pokračuje ligáciou ďalšej sondy na základe komplementarity informatívneho dinukleotidu a opäť sa zaznamená fluorescencia (obr., C, D). Tento cyklus ligácií sa opakuje 5 až 7-krát, takže sa v jednom behu prečíta 25 ((8 – 3) × 5) alebo 35 báz ((8 – 3) × 7) (obr., E).
Výsledkom jedného cyklu ligácií je prečítanie dinukleotidov, medzi ktorými sú vždy 3 bázy neznáme (obr., E). Ako však zistíme, ktorý nukleotid je na akej pozícii, keď rovnakú farbičku majú až 4 kombinácie dinukleotidov? Slúži na to krok nazývaný „reštart“ (denaturácia), kedy do reakcie vstupuje nový univerzálny primer, skrátený o 1 nukleotid z 3'-konca (obr., F). K nemu opäť ligujú sondy ako v prvom kroku, ale zároveň dochádza k prekryvu dinukleotidov z prvého cyklu (obr., G). V tomto kroku získavame dodatočnú informáciu, pretože vieme, akou bázou začína dinukleotid prvej sondy (keďže je zo známej sekvencie kompatibilnej k adaptoru).

Aby bola prečítaná celá sekvencia, je potrebný reštart 4-krát, vždy s univerzálnym primerom skráteným o 1 nukleotid (prvý univerzálny primer je n, po reštartoch je dĺžka a sekvencia primerov n-1, n-2, n-3, n-4). Z obrázka dole vyplýva, že poradie hybridizačných krokov neurčuje zároveň aj poradie čítania nukleotidov v sekvencii, ale sekvencia DNA fragmentu sa začína čítať univerzálnym primerom 2 a pokračuje primermi 1, 5, 4 a končí cyklom s primerom 3. Dekódovanie sekvencie je tak určené práve farebným kódom a sekvenciou prvej bázy pri čítaní univerzálnym primerom 2 (t. j. n-1).


Z uvedeného zložitého postupu vyplýva, že každý nukleotid je fyzicky prečítaný dvakrát, čo výrazne zvyšovalo presnosť tejto metódy (až na 99,94 %). Dvojnásobné čítanie nukleotidov a využitie farebného kódu taktiež umožňovalo excelentnú identifikáciu bodovej mutácie (polymorfizmu), čo bola obrovská výhoda oproti ostatným vtedajším technológiám II. generácie, ktoré mali s vyhodnotením polymorfizmu značné problémy.

Ďalšou výhodou bola relatívne nízka cena (v tom čase 0,13 $ / 1 megabázu). Zásadnou nevýhodou, pre ktorú technológia neprežila, však bolo čítanie len veľmi krátkych sekvencií (len 2 × 25 alebo 2 × 35 báz) a extrémne pomalý beh prístroja, ktorý v závislosti od veľkosti DNA knižnice trval až 8 dní.
III. generácia sekvenovania link
Kým technológie s krátkymi čítaniami (II. generácia, napr. Illumina) dominujú v cene a priepustnosti, ich zásadnou nevýhodou je, že z krátkych úsekov nedokážu správne poskladať zložité repetitívne oblasti DNA. Tento problém vyriešila III. generácia sekvenovania, ktorá sa zamerala na čítanie dlhých úsekov (desiatky tisíc až milióny bázových párov) priamo z jedinej molekuly DNA v reálnom čase, bez nutnosti jej predchádzajúceho namnoženia (amplifikácie PCR). Vďaka tomu nedochádza k chybám a skresleniam spôsobeným polymerázou počas prípravy knižnice.
Práve technológie III. generácie umožnili vedcom po desaťročiach konečne poskladať zložité repetitívne úseky (napr. centroméry a teloméry) a v roku 2022 vďaka nim vedecké konzorcium publikovalo prvú skutočne 100 % kompletnú sekvenciu ľudského genómu bez akýchkoľvek medzier. Dnes sú tieto technológie absolútnym štandardom pre de novo sekvenovanie doteraz neznámych organizmov.
PacBio (Pacific Biosciences) a SMRT sekvenovanie link
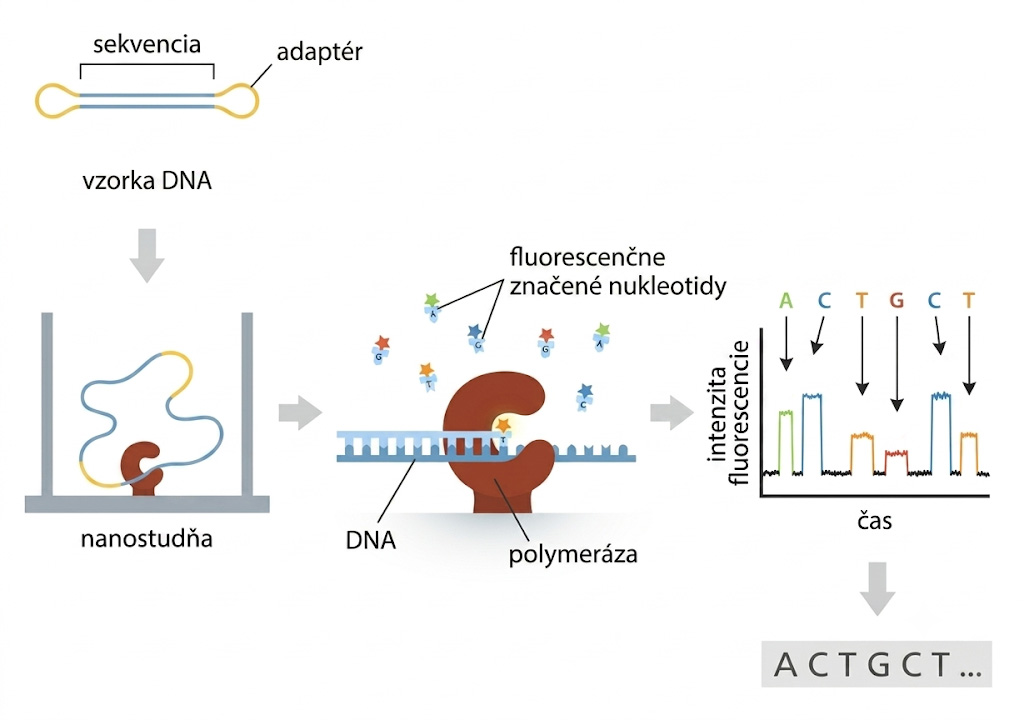
Technológia SMRT (angl. Single Molecule Real Time Sequencing) bola komercionalizovaná v roku 2011. Jej princípom je sledovanie jedinej molekuly DNA-polymerázy, ktorá syntetizuje nové vlákno DNA zloženej do kruhu v mikroskopickej jamke (tzv. ZMW, angl. Zero-Mode Waveguide). Prístroj v reálnom čase opticky zaznamenáva svetelné záblesky z inkorporovaných fluorescenčne značených nukleotidov.

Oxford Nanopore Technologies (nanopórové sekvenovanie) link
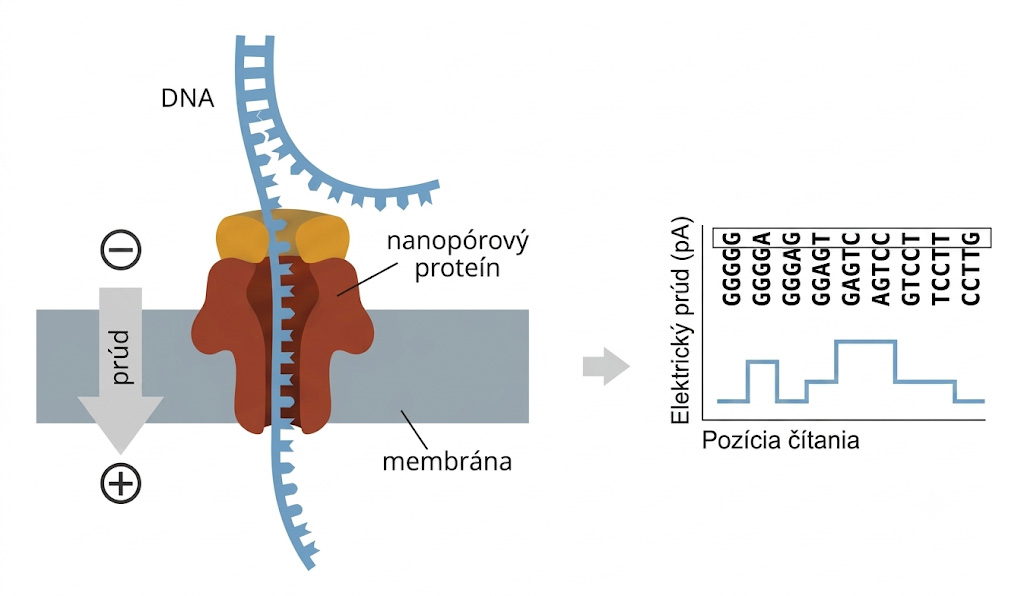
Tento revolučný prístup úplne upustil od optickej detekcie a syntézy nového vlákna. Molekula DNA je pomocou špeciálneho motorického enzýmu doslova „preťahovaná“ cez miniatúrnu bielkovinovú dierku (nanopór) umiestnenú v syntetickej membráne. Pri prechode rôznych nukleotidov (A, C, T, G) cez pór dochádza k charakteristickým zmenám elektrického prúdu, ktoré prístroj meria a analyzuje. Výhodou je možnosť čítať extrémne dlhé fragmenty (tzv. ultra-long reads, presahujúce aj milión bázových párov) a neuveriteľná kompaktnosť prístrojov – základné sekvenátory (napr. MinION) majú veľkosť hrubšej fixky a zapájajú sa do USB portu bežného počítača.
Historický prehľad sekvenačných technológií link
Nasledujúca tabuľka odráža historický stav v čase prechodu z I. na II. generáciu (údaje z obdobia okolo roku 2012). Z dnešného pohľadu sú viaceré uvedené technológie mŕtve (454, SOLiD) a parametre prežívajúcich technológií (najmä cena, dĺžka čítania a dátový výstup) sú neporovnateľne lepšie. Tabuľku ponechávame ako ukážku vtedajšieho technologického skoku a na porovnanie s klasickou Sangerovou metódou.
| 454 GS FLX | HiSeq 2000 | SOLiDv4 | Ion 318 Chip v2 | Sanger 3730xl | |
| sekvenácia | pyrosekvenovanie | sekvenovanie reverzibilnou termináciou syntézy | ligácia sond | sekvenovanie syntézou, zmena pH | dideoxy-nukleotidy, syntéza rôzne veľkých molekúl |
| dĺžka čítania | do 1000 bp | do 150 bp | 2×35 | 200 bp | 400−900 bp |
| presnosť | 99,9 % | 98 % | 99,94 % | 99 % | 99,999 % |
| počet behov | 1 M | 3 G | 1200-1400 M | 4-5,5 M | 1 / kapilára |
| dátový výstup | 0,7 GB | 600 GB | 120 GB | 0,6−1 GB | 1,9−8,4 kB |
| čas behu | 24 hod | 3−10 dní | 7 dní | 2 hod | 20 min - 3 hod |
| cena za 1 Mbp | 10 $ | 0,07 $ | 0,13 $ | 1 $ | 2 400 $ |
Genomika a metagenomika link
Okrem de novo sekvenovania celých genómov vyšších organizmov nachádzajú technológie II. a III. generácie široké uplatnenie pri sekvenovaní menších genómov, napríklad nových bakteriálnych či vírusových kmeňov za účelom analýzy mutácií. Metagenomika, ktorá identifikuje nové mikroorganizmy z rôznych prostredí (vrátane črevnej mikroflóry), sa stala vďaka NGS už rutinnou záležitosťou.
Obrovským boomom posledných rokov sa stala analýza tzv. environmentálnej DNA (eDNA). Vďaka extrémnej citlivosti sekvenátorov dnes dokážu ekológovia a zoológovia presne identifikovať prítomnosť konkrétnych živočíchov a rastlín v ekosystémoch len na základe sekvenovania vzorky vody alebo pôdy, bez akejkoľvek nutnosti fyzického odchytu organizmov.
Klinická prax a personalizovaná medicína link
Kým v minulosti bolo sekvenovanie ľudského genómu pre bežnú diagnostiku pridrahé, dnes už umožňuje rutinnú identifikáciu markerov a predispozícií na multifaktoriálne ochorenia (ako sú cukrovka, vysoký krvný tlak či Alzheimerova choroba). Každá choroba má postupne vytváraný svoj genetický profil, vďaka čomu vieme pacientom stanoviť pravdepodobnosť jej výskytu a včas jej predchádzať.
Obrovskou revolúciou v onkológii sa stali tzv. tekuté biopsie. Vďaka hĺbkovému sekvenovaniu dokážeme dnes zachytiť stopy cirkulujúcej nádorovej DNA (ctDNA) priamo z bežného odberu krvi pacienta, čo umožňuje neinvazívnu diagnostiku rakoviny už v jej úplných zárodkoch.
Napriek technologickej dostupnosti však netreba zabúdať na etické problémy a hrozbu spoločenskej či pracovnej diskriminácie plynúcu z dostupnosti kompletnej genetickej informácie človeka.(13)
Transkriptomika a epigenetika link
Sekvenovanie sa zďaleka neobmedzuje len na DNA. Prepísaním mRNA do cDNA je dnes bežné sekvenovať celé transkriptómy (tzv. RNA-Seq) a zistiť tak presný génový expresný profil tkanív či nádorov, čo úplne nahradilo staršie, zdĺhavé a menej informatívne metódy ako northern blot či microarray čipy.
Kým donedávna sa takto analyzovala len zmes buniek ako celok, moderným štandardom sa stalo tzv. single-cell sekvenovanie, ktoré dokáže prečítať génovú expresiu v každej jednej bunke samostatne.
Rutinne sa už dnes študuje aj epigenetika, napríklad metylácia DNA ako kľúčový regulačný faktor génovej expresie.
- Hajnal, M: Metodika bioinformatickej analýzy dát zo sekvenovania novej generácie typu RNA-seq. (2013). Fakulta informatiky, Masarykova Univerzita. Bakalárska práca.
- Glaus, P: Sekvenovanie génov očami informatika. The University of Manchester.
- Next Generation Sequencing: An Overview - Axygen Biosciences.
- Golstein, C.; Caboche, M: Next-generation high-throughput sequencing technologies. (2008). Technologies of the Future, INRA.
- Wikipedia Contributors: 454 Life Sciences. (2023). Wikimedia Foundation. Wikipedia heslo o priekopníckej technológii pyrosekvenovania.
- Groom, A: Pyrosequencing. Insitute of Genetic Medicine, New Castle University.
- Shackelford, R. E: Molecular Pathology: DNA sequencing: Pyrosequencing. (2011). Tulane University.
- Illumina Sequencing Technology. (2010). Illumina.
- Ion PGM System Specifications.
- Wang, Y: SOLiD Bioinformatics Overview. (2009). Applied Biosystems, Life Technologies.
- Liu, L., et al: Comparison of next‐generation sequencing systems. (2012). BioMed Research International. 2012. 251364. DOI: 10.1155/2012/251364.
- Vídeňská, P: Next generation sequencing. MikroDok.
- National Human Genome Research Insitute. DNA Sequencing Costs.


